
Overcoming Dynamic Range in MS-Based Blood Proteomics: Depletion and Enrichment Methods
Human blood plasma is notoriously complex, containing proteins across an extremely wide concentration range-from major proteins at ~35–50 mg/mL to trace proteins in the picogram-per-milliliter range. In fact, the ten most abundant plasma proteins (led by albumin and immunoglobulins) account for roughly 90% of total protein mass. This vast dynamic range means that highly abundant proteins can mask the detection of low-abundance biomarkers in MS-based proteomic analyses. For pharmaceutical, biotech, and academic researchers aiming to discover disease biomarkers or drug targets, overcoming this masking effect is crucial.
To address this, specialized mass spectrometry sample preparation techniques have been developed to either deplete high-abundance proteins or enrich low-abundance proteins in plasma/serum prior to analysis. By reducing sample complexity, these methods make it easier to detect and quantify proteins present at ng/mL to pg/mL levels. Below, we introduce the most commonly used plasma proteomics methods-from immunoaffinity columns to novel nanoparticle reagents-and compare how each improves proteome coverage. We focus on human plasma/serum workflows and highlight recent advancements (past 5 years) relevant to clients in pharma, biotech, and academic research.
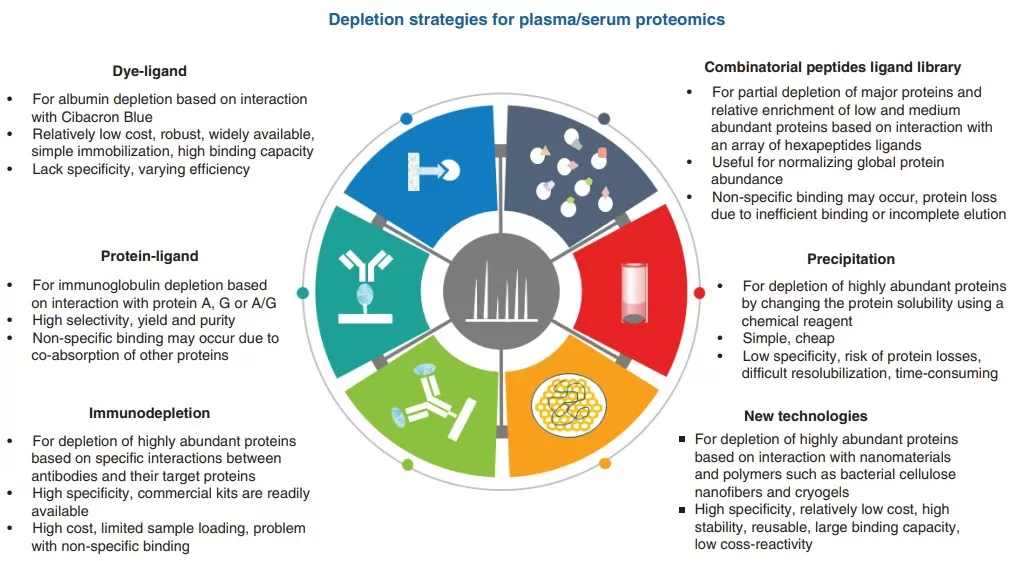
Different depletion methods used to remove highly abundant proteins in plasma/serum (Lee et al., 2019)
Low-Abundance Protein Enrichment Using Nanoparticles
How It Works: Nanoparticle-based enrichment is a powerful recent advancement in plasma proteomics. This approach exploits the natural tendency of nanoparticles (NPs) to adsorb proteins from biofluids, forming what’s known as a protein corona on their surfaces. By engineering NPs with different surface chemistries, charges, or pore structures, distinct subsets of plasma proteins preferentially bind to different nanoparticles, effectively partitioning the proteome based on physicochemical traits. Unlike antibody capture, this is broadly unbiased—NPs do not rely on predefined targets, allowing a more comprehensive sampling of the proteome. This innovative technology has already been adopted by leading proteomics service platforms, including MetwareBio’s blood proteomics platform and the Seer Proteograph, to achieve deep and reproducible plasma profiling. In practice, plasma is incubated with a diverse panel of nanoparticles (such as silica, polymer, or iron oxide cores with various coatings). Each type of particle forms a corona with a unique fraction of plasma proteins. The nanoparticles are then magnetically separated, washed, and the bound proteins digested into peptides for LC-MS analysis. Because each NP type has a limited capacity and selective affinities, high-abundance proteins may saturate certain nanoparticles but leave others free to capture mid- and low-abundance proteins, resulting in a “democratized” proteome representation that enhances low-copy protein detection.

Formation of NP protein corona (Blume et al., 2020)
Pros and Cons: The primary advantage of nanoparticle enrichment is its exceptional breadth and depth, effectively addressing plasma’s vast dynamic range. This strategy has demonstrated the ability to identify thousands of proteins per sample. For instance, MetwareBio’s platform routinely detects over 4,000 proteins in human plasma, empowering pharmaceutical and clinical researchers to discover critical low-abundance biomarkers. It is also well-suited for high-throughput studies. However, while broadly unbiased, each magnetic nanoparticle formulation does have intrinsic binding preferences, meaning certain protein classes may be enriched over others. Despite this, extensive validation shows excellent reproducibility and robust quantitative performance, making nanoparticle-based enrichment a next-generation solution for low-abundance protein enrichment and comprehensive mass spectrometry sample preparation in blood proteomics.

Number of proteins identified in plasma/serum samples from various of species via MetwareBio’s blood proteomics platform
Immunoaffinity Depletion of High-Abundance Proteins
How It Works: Immunoaffinity depletion uses antibodies to selectively bind and remove specific high-abundance proteins from plasma. Commercial columns like Agilent’s MARS or Sigma’s Seppro are packed with immobilized antibodies (often polyclonal IgG or IgY) targeting a set of major proteins (e.g. albumin, IgG, transferrin, IgA, haptoglobin, etc.). When diluted plasma is passed through the column, the target proteins bind and are retained, while the rest of the proteome flows through for collection. Typically, kits are available to remove Top-2, Top-6, Top-12 or even Top-20 abundant proteins. For example, the ProteoPrep 20 kit immunodepletes the 20 highest abundance proteins, eliminating ~97% of total plasma protein mass. Removing this “protein clutter” allows a much greater volume of plasma to be loaded into an LC–MS workflow, thereby boosting sensitivity for low-copy proteins. In essence, immuno-depletion narrows the dynamic range by subtracting the dominant proteins.
Pros and Cons: The clear advantage of immunoaffinity columns is specificity-they very effectively and reproducibly remove known high-abundance interferents. This enables significant proteome depth gains; studies show that multi-protein depletion can reveal ~25% more proteins in a plasma sample compared to no depletion. In practical terms, by taking out ~90–99% of total protein, one can inject correspondingly more sample or detect lower-level peptides that were previously co-eluting with abundant proteins. Immuno-depletion is a well-established, kit-available approach (e.g. Agilent MARS6/MARS14, Thermo Pierce Top12, Sigma Seppro IgY columns) that fits easily into LC-MS workflows. However, the method comes with trade-offs. The antibody columns and spin cartridges are relatively expensive and typically single-use or limited-use. They also have finite binding capacity-only ~10–20 μL of plasma per spin column in many cases-so processing large sample volumes may require multiple runs. Another limitation is the potential loss of certain low-abundance proteins that bind to or co-elute with the targeted abundant proteins. For example, albumin is a carrier for many small peptides; depleting albumin might unintentionally remove those bound peptides (though manufacturers optimize buffers to minimize non-specific binding). In summary, immunoaffinity depletion offers a highly targeted way to reduce plasma complexity, with strong performance gains in downstream analysis, but at a relatively high cost and a risk of losing any biomarker that happens to be captured along with the depleted proteins.
Combinatorial Ligand Library Enrichment
How It Works: An alternative to physically removing abundant proteins is to down-size their representation while pulling up the rarer proteins-this is the idea behind combinatorial peptide ligand libraries. The most well-known implementation is Bio-Rad’s ProteoMiner kit, which uses a solid-phase library of millions of hexapeptides attached to beads. These random peptide ligands collectively have binding affinity for a very wide array of proteins. When plasma is incubated with the beads, high-abundance proteins quickly saturate their few complementary peptide ligands, and excess high-abundance molecules remain unbound (and are washed away). In contrast, low-abundance proteins are far from saturating any ligands, so virtually all molecules of those proteins in the sample can bind to the beads. In effect, ProteoMiner “equalizes” the protein concentrations: abundant proteins get diluted (by limited binding capacity), while rare proteins get concentrated on the beads. After washing, the bound proteins (enriched in low-abundance species) are eluted or on-bead digested for analysis. This combinatorial enrichment approach doesn’t target specific proteins, but rather broadly compresses the dynamic range of the sample.
Pros and Cons: The combinatorial ligand library approach is broad and unbiased, theoretically capturing proteins without needing prior target knowledge. This enables discovery of novel low-abundance proteins often missed in untreated plasma or even after immunodepletion. For example, ProteoMiner enrichment has revealed plasma proteins at ~10 pg/mL levels that would otherwise go undetected. It’s also relatively straightforward (mix-incubate-spin) and more cost-effective per sample than antibody columns. Importantly, larger plasma volumes (e.g., ≥100 µL) can be processed since bead capacity, not sample size, is the limiting factor—allowing more total plasma to be mined for low-abundance biomarkers. However, proteome depth gains are sometimes less than with targeted depletion: one study found a Top-20 immunodepletion column yielded ~25% more unique proteins than ProteoMiner on the same sample. Reproducibility can also vary; since binding relies on random hexapeptides, different lots or runs may enrich slightly different subsets of proteins. Moreover, very abundant proteins are only partially reduced, so samples remain less “clean” than after immunodepletion. Some proteins may lack affinity to any ligands and thus go undetected. In practice, combinatorial ligand libraries are often combined with immunodepletion or fractionation to maximize coverage. Overall, they offer a practical, commercially available strategy to enrich low-abundance proteins, albeit with variable efficiency.
Chemical Precipitation Methods for Depletion
How It Works: Chemical precipitation leverages simple solvent or acid-induced protein aggregation to reduce high-abundance plasma proteins. Adding perchloric acid (HClO₄) or organic solvents like methanol, ethanol, or acetone denatures and precipitates major proteins such as albumin and fibrinogen, which are then removed by centrifugation. The remaining supernatant becomes enriched in lower-abundance proteins and peptides. Recent studies comparing different precipitation methods found methanol yielded the best protein identifications and consistency, routinely achieving over 600 protein groups, with some detected near 10 pg/mL. Perchloric acid treatments also effectively reduced dominant proteins, enhancing low-abundance detection and proving valuable as a quick depletion step.
Pros and Cons: Precipitation’s biggest advantages are its low cost and simplicity—requiring only standard lab chemicals, no specialized kits—making it ideal for high-throughput or large-cohort studies. It also handles relatively large plasma volumes easily. When optimized, it significantly broadens proteome coverage; for example, methanol precipitation enabled detection of 700+ proteins, including many low-abundance candidates. However, this approach is non-specific. Many proteins precipitate together, so interesting mid- and low-abundance proteins might also be lost with albumin. It’s essentially a crude filter that trades some selectivity for ease. Residual solvents or salts may require additional cleanup before MS analysis, and without standardized ratios or incubation times, reproducibility can vary across labs. Nonetheless, precipitation remains a practical, scalable strategy—especially when expensive depletion kits aren’t feasible—offering substantial gains in plasma proteome depth for exploratory mass spectrometry studies.
Is Ultrafiltration Still Relevant for Plasma Proteomics?
How It Works: Ultrafiltration was one of the earliest approaches to enrich low-molecular-weight (LMW) proteins in plasma by physically separating proteins based on size. Using centrifugal filter devices with a defined molecular-weight cutoff (MWCO), plasma can be fractionated into “large” proteins retained by the filter (like albumin and IgG) and “small” proteins and peptides that pass into the filtrate. For instance, a 30 kDa or 50 kDa filter will hold back most major plasma proteins while allowing smaller molecules to flow through. Some protocols even use sequential filters (100 kDa → 50 kDa → 10 kDa) to create multiple size-based fractions. The goal is to access potential biomarkers in the LMW proteome, such as cytokines, hormones, or proteolytic fragments, with less interference from dominant high-mass proteins.
Pros and Cons: Ultrafiltration’s main advantages are its simplicity (just spin through a membrane) and the absence of costly reagents. It’s also unbiased in that size is the only selection factor, making it broadly applicable. Studies have shown ultrafiltration can reveal unique LMW proteins not detected in whole plasma, potentially capturing small biomarkers missed by immunodepletion or precipitation. However, it has notable drawbacks. Size alone doesn’t guarantee clean separation—many peptides in the <30 kDa fraction are actually fragments of high-abundance proteins generated by natural proteolysis, so the filtrate can still be dominated by albumin or fibrinogen fragments rather than novel low-level targets. Additionally, filters can clog, leading to inconsistent yields, and proteins may adhere to the membrane, reducing recovery. As a result, filtrates often show only modest increases in detectable proteins (sometimes just dozens more), limiting impact on overall proteome depth. Recent literature suggests ultrafiltration is best suited for niche applications like peptidomics or hormone studies, rather than comprehensive plasma proteomics. Today, it’s often combined with other enrichment steps or replaced by more advanced strategies. While straightforward, ultrafiltration requires careful execution to minimize losses and is generally not the first-line choice for deep plasma profiling.
Summary Comparison of Depletion/Enrichment Methods
To wrap up, the table below provides a side-by-side comparison of the key attributes of each method discussed:
|
Method |
Specificity |
Proteome Depth Gain |
Cost |
Commercial Availability |
Ease of Use |
|
Nanoparticle Enrichment (protein coronas) |
Moderate/Broad-different NP surfaces selectively bind subsets of proteins |
Very High-thousands of proteins can be identified (2000–3000 in studies) by capturing wide dynamic range |
High-emerging tech (specialized nanoparticles and instruments) |
Limited but growing (e.g. Seer Proteograph platform; some research-grade NP materials) |
Moderate-protocol is automatable, but currently requires specific kits/instruments |
|
Immunoaffinity Depletion (antibody columns) |
High–targets specific known high-abundance proteins |
High-allows ~20–50× more low-abundance proteins to be detected |
High-antibody columns and kits are expensive |
Widely available kits (MARS, Seppro, ProteoPrep) |
Moderate-straightforward protocol but limited sample load per run |
|
Combinatorial Ligand Library (ProteoMiner beads) |
Low/Broad-binds a wide range of proteins via random ligands |
Moderate-enhances low-abundance detection; ~75% of proteins vs immunodepletion in one study |
Moderate-kit cost is lower than antibody columns |
Available (Bio-Rad ProteoMiner kit); technique used in research |
Moderate-simple bead incubation, but some variability in results |
|
Chemical Precipitation (acid/solvent) |
Low-non-specific precipitation of many proteins together |
Moderate-significantly reduces major proteins; >600–700 proteins identified in resultant sample |
Low-uses inexpensive reagents (methanol, acids) |
No special kit needed; standard protocols in literature |
High-easy, quick steps; suitable for high-throughput labs |
|
Ultrafiltration (molecular weight cutoff) |
Moderate-size-based specificity (e.g. <30 kDa fraction) |
Low/Targeted-enriches small proteins / peptides; tens of unique LMW proteins, but many fragments of bigger ones |
Low-uses disposable filter units (relatively inexpensive) |
Broadly available (Amicon/Millipore ultrafiltration devices) |
Moderate-simple to perform, but careful protocol needed to avoid filter clogging or loss |
Each approach has unique strengths. Immunoaffinity depletion offers targeted removal of known interferents and is a trusted method for improving plasma mass spectrometry results, ideal when sample quantity is limited and budget allows. ProteoMiner and CPLL enrichment cast a wide net to bring up the “long tail” of the proteome, useful in discovery-phase projects. Precipitation methods are attractive for their cost-effectiveness and surprising performance gains – a good choice for initial screens or when scaling up to hundreds of samples. Ultrafiltration serves niche purposes, particularly in peptidomics, but is less commonly the sole method in modern plasma proteomics workflows. Finally, nanoparticle-based strategies represent the cutting edge, pushing the depth of plasma protein detection to new heights, albeit with higher complexity and cost. In practice, these methods can also be combined (for example, depleting top proteins, then enriching low ones) to further enhance results. By understanding the pros and cons of each technique, researchers in pharma, biotech, and academia can choose the optimal sample preparation strategy for their blood proteomics studies – balancing depth, throughput, and practical considerations to unlock the low-abundance proteome for biomarker and therapeutic discovery.
References
1. Rice, Shawn J., et al. “Characterization of Effective, Simple, and Low-Cost Precipitation Methods for Depleting Abundant Plasma Proteins to Enhance the Depth and Breadth of Plasma Proteomics.” Proteomics, vol. 24, no. 15, 2024, e2400071.
2. Li, Nan et al. “The Development of a Novel Zeolite-Based Assay for Efficient and Deep Plasma Proteomic Profiling.” Journal of Nanobiotechnology, vol. 22, no. 1, 2024, article 164.
3. Blume, John E., et al. “Rapid, Deep and Precise Profiling of the Plasma Proteome with Multi–Nanoparticle Protein Corona.” Nature Communications, vol. 11, 2020, article 3662.
4. Lee, Pey Yee, et al. “Plasma/Serum Proteomics: Depletion Strategies for Reducing High-Abundance Proteins for Biomarker Discovery.” Bioanalysis, vol. 11, no. 19, 2019, pp. 1799–1812.
5. Millioni, Renato, et al. “High-Abundance Proteins Depletion vs Low-Abundance Proteins Enrichment: Comparison of Methods to Reduce the Plasma Proteome Complexity.” PLoS ONE, vol. 6, no. 5, 2011, e19603.
6. Greening, David W., and Richard J. Simpson. “A Centrifugal Ultrafiltration Strategy for Isolating the Low-Molecular-Weight (≤25K) Component of the Human Plasma Proteome.” Journal of Proteomics, vol. 73, no. 3, 2010, pp. 637–648.
Next-Generation Omics Solutions:
Proteomics & Metabolomics
Ready to get started? Submit your inquiry or contact us at support-global@metwarebio.com.
