
Data Calibration in TMT/iTRAQ Proteomics: How to Identify and Correct Channel Interference and Ratio Compression
Tandem Mass Tag (TMT) and Isobaric Tags for Relative and Absolute Quantification (iTRAQ) are leading techniques in label-based quantitative proteomics, enabling high-throughput protein identification and quantification. These methods allow researchers to precisely analyze protein expression across multiple samples, making them essential for applications like biomarker discovery, disease mechanism research, and drug development. However, despite their power, TMT/iTRAQ quantification can still be affected by certain technical challenges that undermine data accuracy. (Learn more at: Label-based Protein Quantification Technology—iTRAQ, TMT, SILAC)
Two major issues—channel interference and ratio compression—can significantly affect the reliability of TMT/iTRAQ data. These problems can lead to the underestimation of protein differences or misidentify proteins as differential when no real change exists, potentially skewing research conclusions. Understanding the underlying causes of these errors and implementing effective calibration strategies is crucial to improving data accuracy.
In this blog, we will discuss the nature of these issues, their impact on quantitative results, and calibration techniques such as internal reference calibration and mixed sample calibration that can correct these biases. With a better understanding of these challenges and solutions, you'll be equipped to improve the accuracy and reliability of your label-based proteomics experiments.
Introduction to TMT/iTRAQ: Workflow and Quantification Principles
Tandem Mass Tag (TMT) and Isobaric Tags for Relative and Absolute Quantification (iTRAQ) are advanced technologies developed by Thermo Fisher Scientific and AB Sciex, respectively, to provide high-throughput, label-based quantitative proteomics. These platforms have transformed the field by enabling precise quantification of proteins across multiple biological samples in a single experiment. However, to fully understand the challenges that can arise in TMT/iTRAQ data analysis, it is important to first explore the fundamental workflow and the quantitative principles that underpin these methods.
TMT/iTRAQ Experimental Workflow
The TMT (Tandem Mass Tag) and iTRAQ (Isobaric Tags for Relative and Absolute Quantification) technologies are based on isobaric labeling, which allows for the simultaneous quantification of multiple samples in a single mass spectrometry experiment. The workflow typically consists of the following steps:
1) Sample Preparation: Proteins are extracted from biological samples, digested with enzymes (usually trypsin), and the resulting peptides are labeled with isobaric tags such as TMT or iTRAQ. Each tag is assigned to a specific sample, allowing multiple samples to be pooled together for analysis.
2) Labeling: The TMT/iTRAQ tags, which are isobaric in nature, are designed to have the same mass. Each tag contains a unique reporter ion, which is released during mass spectrometry analysis and correlates to the abundance of peptides from each sample.
3) Peptide Separation: The labeled peptides are separated using liquid chromatography (LC) to reduce complexity and ensure better resolution during MS analysis.
4) Mass Spectrometry Analysis: The peptides are analyzed using tandem mass spectrometry (MS/MS), where precursor ions are fragmented, and reporter ions are quantified to determine peptide abundance. The mass spectrometer detects the reporter ions to calculate the relative abundance of peptides in each sample.
5) Data Analysis: The acquired data is processed and analyzed, where the intensity of the reporter ions is used to quantify protein abundance. Statistical methods are applied to identify differentially expressed proteins and assess their biological significance.
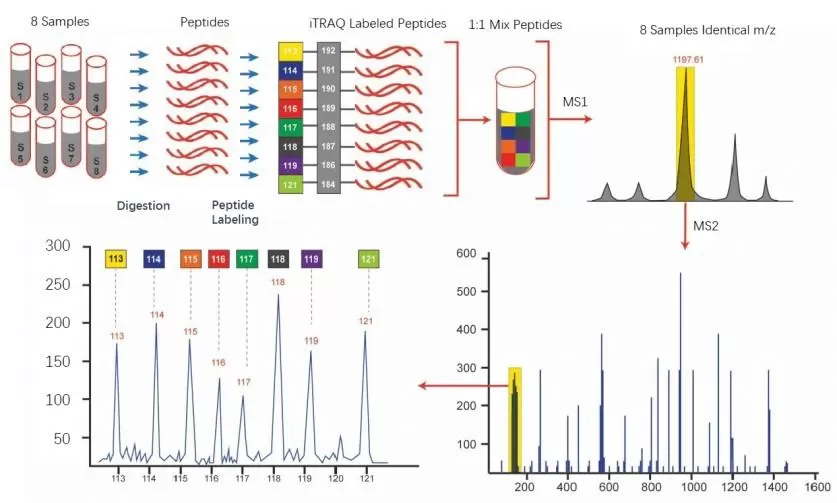
The workflow of iTRAQ analysis
Quantitative Principles of TMT/iTRAQ
The core principle behind TMT/iTRAQ quantification is the use of isobaric tags, which are chemically identical except for their isotopic mass. This allows multiple samples to be analyzed together in a single experiment without the need for separate measurements. The quantification is based on the intensity of the reporter ions released during MS/MS fragmentation, which are unique to each tag.
i. Isobaric Tagging: Each TMT or iTRAQ tag consists of a unique reporter group and a balance group that ensures the overall mass remains constant. During MS/MS fragmentation, the reporter group is cleaved from the peptide and detected as an ion. The intensity of this reporter ion reflects the abundance of the peptide in the corresponding sample.
ii. Relative Quantification: The quantitative information is derived from comparing the reporter ion intensities across samples. Higher intensity indicates greater abundance of the peptide in the sample. These ratios are used to determine relative protein expression levels between the different samples analyzed together.
iii. Absolute Quantification: In some cases, absolute quantification can also be performed by using a standard peptide with a known concentration. The intensity of the reporter ion from the standard peptide can be compared to the intensity from the target peptides to estimate their absolute abundance.
This label-based quantification method allows for accurate, high-throughput protein analysis, making TMT/iTRAQ ideal for comparing protein expression across multiple experimental conditions, such as different time points, treatments, or disease states.

The structure of Itraq reagents
Technical Challenges in TMT/iTRAQ: Channel Interference and Ratio Compression
Despite the widespread use of TMT and iTRAQ in label-based quantitative proteomics, two longstanding and well-recognized challenges continue to affect the accuracy of data: channel interference and ratio compression. These issues have been consistently observed across numerous studies and are inherent to the nature of isobaric tagging techniques. Both problems arise from distinct causes but are fundamentally linked by "signal contamination" or "imbalance in ratios," which disrupt the integrity of quantitative measurements.
Channel Interference: Signal Cross-Contamination Across Channels
Channel interference can be likened to a "cross-contamination" at the signal level. It occurs when signals from different labeled channels merge, causing the signal from one channel to include non-target signals from another, leading to an inaccurate quantitative result. The primary causes of channel interference are:
- Inherent Defects in Labeling Reagents
Ideally, TMT/iTRAQ labels differ only in isotopic composition, while other physicochemical properties should be identical. However, during production, slight impurities, such as isotopic contamination, are inevitable. For example, the TMT 126 channel label may contain a small amount of TMT 127 isotopes. These "impurity signals" directly contaminate adjacent channels, particularly in the detection of low-abundance peptides, where impurity signals become more prominent, exacerbating cross-talk problems.
- Co-elution Interference During Sample Separation and Detection
Complex biological samples (e.g., tumor tissues, blood) contain thousands of peptide species. During liquid chromatography, some peptides with similar physicochemical properties cannot be completely separated and co-elute into the mass spectrometer's fragmentation window. When these co-eluting peptides are fragmented, they release reporter ions from different channels, resulting in signal overlap. For instance, if peptide segments from sample A and sample B co-elute, their signals will be detected simultaneously, leading to deviations in both channels. This interference becomes more pronounced when sample complexity is high.
Ratio Compression: The Underestimation Trap of Differential Abundance
If channel interference is "signal contamination," ratio compression is "signal shrinkage." The observed protein abundance ratios are often significantly lower than the true differences, leading to the omission of low-abundance differential proteins and increasing the false-negative rate. The core cause lies in the "co-isolation and co-fragmentation effect," which can be broken down into three components:
- Dilution Effect of Background Peptides
The mass spectrometer's fragmentation window is typically set to a certain width, which means that besides the target peptides, background peptides also enter the window. The non-specific signals from these background peptides dilute the true signal of the target peptides. For example, if the true difference in a peptide's abundance between two samples is 10-fold, the observed difference might only be 4–6-fold due to the dilution caused by background signals.

Experimental design for evaluating the interference in TMT29 [2].
(A) Schematic representation of TMT11 and TMT18 samples in TMT29 analysis. Digested E. coli and human peptides were TMT-labeled and mixed at known ratios in the TMT11 and TMT18 sets. These two sets were then pooled together at 1:1 ratio (human peptides) in each channel with the backgrounD and analyzed by LC-MS/MS. (B–E) The averaged experimental relative intensities in TMT29. (B, C) The experimental relative intensities without human background (interference) which are closed to theoretical values. The experimental relative intensities in (D, E) are heavily distorted by interference derived from the human background.
- Labeling Efficiency and Sample Mixing Errors
If the peptide labeling efficiency varies across different samples (e.g., some samples are incompletely labeled, leaving behind unlabeled peptides), the unlabeled peptides generate non-specific signals, exacerbating ratio compression. Additionally, improper control of volume and concentration during sample mixing after labeling can lead to deviations from the ideal initial ratio, which prevents the observed ratio from accurately reflecting the true differences.
- Competition for Signal Resources and Noise Flattening
During the limited detection time, high-abundance peptide fragment ions consume most of the signal collection resources. Coupled with shared chemical background noise across all channels, extreme ratios are "pulled" toward the middle value, leading to the underestimation of the difference.
Key Indicators of Channel Interference and Ratio Compression
Due to the inherent nature of channel interference and ratio compression in TMT/iTRAQ experiments, these issues are often unavoidable, leading to data distortions. To ensure data reliability, it’s essential to identify these problems early. Specific signs, such as unusual channel correlations or unexpected changes in protein ratios, serve as key indicators of data abnormalities, allowing for timely intervention before they compromise analysis.
1) The most common "warning signs" of channel interference include:
- Abnormal Blank Control Signals: If the signal intensity from the blank control channel (e.g., unlabeled peptide channels) exceeds the background noise by more than three times.
- High Channel Correlation: If the signal correlation between sample channels without biological relationship (R² > 0.8) is high, this indicates signal contamination.
- Deviation in Standard Ratios: If a known internal standard peptide with a known concentration ratio is added, and the detected ratio deviates from the theoretical ratio by more than 20% (excluding labeling efficiency issues).
2) Typical signs of ratio compression include:
- Non-Linear Standard Curve: When the standard protein undergoes gradient dilution, and the observed ratio does not change linearly with theoretical concentrations, but instead "flattens."
- Low Differential Fold Change: When expected significant differences between samples (e.g., drug-treated vs. control) show fold changes of only 1.2–1.5, lacking proteins with large fold changes.
- Impure MS² Spectra: When the fragment ion peaks of target peptides make up less than 70% of the total peak, with many additional peaks.
- Poor Reproducibility: In repeated experiments, the fold change of the same differential protein fluctuates significantly, with operational errors unable to explain the variations.
Calibration Algorithms for Channel Interference and Ratio Compression
To address channel interference and ratio compression, several robust calibration algorithms have been developed to correct for these problems and improve the accuracy of quantitative results. Among them, internal reference calibration and mixed sample calibration have emerged as the most widely used techniques due to their straightforward implementation and adaptability across different experimental setups. These methods help mitigate the effects of signal contamination and imbalance, ensuring that the data reflects true protein abundance levels, thus enhancing the reliability of proteomics analysis.
Internal Reference Calibration
Internal reference calibration works by introducing an internal reference protein (or peptide) with a known concentration into each sample. Using its theoretical ratio as a baseline, a calibration model is built to correct the measured ratios of all proteins detected. This method simultaneously corrects for both channel interference and ratio compression, with low thresholds and stable results. The internal reference must meet the criteria of "stable expression across all samples, moderate abundance, and similar physicochemical properties to target proteins." After adding equal concentrations of internal reference to each sample, ideally, the measured ratio of the internal reference across all channels should be close to 1 (relative quantification) or equal to the theoretical added ratio (absolute quantification). If the measured ratio deviates from the theoretical value, the correction factor is calculated as the "theoretical ratio / measured ratio" and applied to all protein ratios.
Calibration Based on Mixed Samples
Mixed sample calibration (also known as "reference channel calibration") works by mixing all the samples to be tested in equal proportions to create a "mixed reference sample." After labeling this mixed sample with a single channel, its signal serves as a "global reference" to correct the deviations in other channels. This method can simultaneously correct for channel interference, ratio compression, and batch biases, with higher calibration accuracy. Since the mixed reference sample reflects the average abundance level of all the samples, it remains consistent across different batches and can serve as a stable reference. If the signal from a channel deviates too much from the mixed reference channel, a global correction model can be established based on the ratio distribution between the mixed reference and the channel to be calibrated, standardizing the signals.
Tips for Choosing Calibration Algorithms
When selecting between internal reference calibration and mixed sample calibration, the decision depends on the experimental design, sample characteristics, required quantification accuracy, cost, and operational complexity. There is no absolute "best" choice—only differences in "adaptability." Internal reference calibration excels in its simplicity, requiring no additional experimental design and is suitable for small samples or targeted quantification. On the other hand, mixed sample calibration excels in eliminating exogenous interference, adapting well to large-scale full-spectrum quantification, and addressing systematic errors across channels/batches more comprehensively.
Future Perspectives: Advancing Accuracy in TMT/iTRAQ Proteomics
The high-throughput and high-sensitivity advantages of TMT/iTRAQ technology have significantly enhanced the efficiency of proteomics research. However, achieving accurate quantification requires overcoming the challenges of channel interference and ratio compression. Internal reference calibration and mixed sample calibration, with their core approaches of "anchoring with known standards" and "global reference standardization," offer mature and reliable solutions for data correction. Calibration algorithms should be complemented by quality control throughout the entire experimental process: ensuring complete sample lysis and consistent enzyme digestion before labeling; controlling reagent volumes and reaction conditions during labeling to achieve labeling efficiency > 95%; optimizing fragmentation window widths during mass spectrometry to reduce co-isolation interference. Only by combining "calibration algorithms" with "process quality control" can deviations be minimized to the greatest extent.
As proteomics technologies continue to evolve, calibration algorithms are progressing toward intelligent upgrades. However, due to their stability and applicability, internal reference calibration and mixed sample calibration remain core choices in laboratories. Mastering the principles and applications of these algorithms ensures that TMT/iTRAQ data reflects its true biological meaning, providing a solid foundation for biomarker discovery, disease mechanism research, and drug development.
Reference
1. Searle, S. J., et al. (2019). An efficient solution for resolving iTRAQ and TMT channel cross-talk. Proteomics, 19(18), e201900243. DOI: 10.1002/pmic.201900243
2. Sun H, Poudel S, Vanderwall D, Lee DG, Li Y, Peng J. 29-Plex tandem mass tag mass spectrometry enabling accurate quantification by interference correction. Proteomics. 2022 Oct;22(19-20):e2100243. doi: 10.1002/pmic.202100243. Epub 2022 Jun 27. PMID: 35723178; PMCID: PMC9588555.
3. Kanashova, T., et al. (2019). TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Molecular & Cellular Proteomics, 18(12), 2409-2420.DOI: 10.1074/mcp.RA119.001727
4. Thermo Fisher Scientific. (2018). Development of a Quality Control Standard for Tandem Mass Tags (TMT) Workflows. ASMS 2018 Poster. PO-65242.
5. Liu, Y., et al. (2020). Assessment of TMT Labeling Efficiency in Large-Scale Quantitative Proteomics: The Critical Effect of Sample pH. Journal of Proteome Research, 19(7), 2946-2955.
6. Currie, E., et al. (2021). Simultaneous proteome localization and turnover analysis reveals spatiotemporal features of protein homeostasis disruptions. Molecular & Cellular Proteomics, 20(12), 100096.
7. Wang, Z., et al. (2020). Multiplexed Data-independent Acquisition-based Proteomics Enabled by TMTpro Complementary Ions. Analytical Chemistry, 92(2), 1607-1615.
8. Searle, S. J., et al. (2019). An efficient solution for resolving iTRAQ and TMT channel cross-talk. Proteomics, 19(18), e201900243.
Next-Generation Omics Solutions:
Proteomics & Metabolomics
Ready to get started? Submit your inquiry or contact us at support-global@metwarebio.com.
